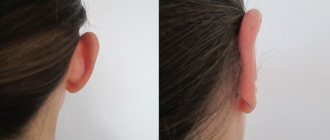
There are 4 degrees of microtia:
Grade I:
A smaller version of the regular ear, with the same physical characteristics and the external auditory canal present.
Grade II:
Partially formed outer ear with a very small or narrow ear canal.
The ear canal may become closed (canal stenosis), resulting in hearing loss. III degree:
Absence of the outer ear of a typical shape (only a small remnant of cartilage and earlobe is present), absence of the external auditory canal and tympanic membrane (auricular atresia).
IV degree:
absence of ear (anotia).
Causes and severity of the anomaly
In male children, compared to girls, microtia and atresia of the external auditory canal are observed 2-2.5 times more often. As a rule, this defect is unilateral and most often localized on the right, but in approximately 10% of cases the pathology is bilateral.
To date, the causes of the pathology have not been established. Many different hypotheses for its development have been proposed, for example, the influence of viruses, in particular the measles virus, damage to blood vessels, the toxic effect on the fetus of various drugs taken by a woman during pregnancy, diabetes, the influence of an unhealthy lifestyle (drinking alcohol, smoking, stressful condition) of a pregnant woman and environmental factors, etc.
However, all these hypotheses did not stand the test - upon further study, none of them were confirmed. The presence of a hereditary predisposition has been proven in some newborns, but this reason is not decisive. Even in families in which both parents have microtia, most often children are born with normal ears and external auditory canals.
In 85% of cases, the disease is sporadic (scattered).
Only 15% are one of the manifestations of hereditary pathology, and of these, half of the cases are bilateral microtia. In addition, the anomaly in question may be one of the manifestations of such hereditary diseases as Konigsmarck, Treacher-Collins, and Goldenhar syndromes.
Bilateral congenital pathology with atresia of the auditory canal without auditory prosthetics from infancy leads to a delay in the development of speech, perception, memory, thinking processes, logic, imagination, the formation of concepts and ideas, etc.
The severity of the abnormal condition varies from a moderate reduction in size and slightly pronounced deformation of the auricle to its complete absence (anotia) and atresia of the external auditory canal. A large number of existing classifications of congenital hearing abnormalities are based on etiopathogenetic and clinical signs. Depending on the severity of the pathology, four degrees are distinguished:
- I degree - the auricle is slightly reduced, the external auditory canal is preserved, but its diameter is slightly narrower compared to the norm.
- II degree - the auricle is partially underdeveloped, the ear canal is very narrow or absent, the perception of sounds is partially reduced.
- III - the auricle is a rudiment and has the appearance of a rudiment, the auditory canal and eardrum are absent, hearing is significantly reduced.
- IV - anotia.
However, the presence of microtia in a newborn is assessed by most plastic surgeons in accordance with H. Weerda’s classification, which reflects the degree of isolated developmental anomaly of the auricle only (without taking into account changes in the auditory canal):
- Microtia of the first degree - the auricle is flattened, bent and ingrown, has smaller dimensions than normal, the earlobe is deformed, but all elements are anatomically slightly changed and are easily recognizable.
- Microtia of the II degree is a small-sized drooping auricle, the upper part of which is represented by an underdeveloped, as if curled, curl.
- Microtia grade III is the most severe form. This is a deep underdevelopment of the ear, manifested in the presence of only rudimentary remains - a skin-cartilaginous ridge with a lobe, only a lobe, or the complete absence of even rudiments (anotia).
Otolaryngologist surgeons often use the H. Schuknecht classification. It most fully reflects the specifics of the developmental anomaly depending on changes in the ear canal and the degree of hearing loss, and helps in choosing treatment tactics. This classification is based on the types of auditory canal atresia:
- Type “A” - atresia is noted only in the cartilaginous part of the external auditory canal. In this case, there is a hearing loss of the first degree.
- Type “B” - atresia affects both cartilaginous and bone parts. Hearing is reduced to II-III degree.
- Type “C” - any form of complete atresia, hypoplasia of the tympanic membrane.
- Type “D” is complete atresia, accompanied by a slight air content (pneumatization) of the mastoid process of the temporal bone, incorrect location of the labyrinthine capsule and the facial nerve canal. Such changes are a contraindication to surgical operations to improve hearing.
Is microtia a genetic disease?
Yes and no.
No:
There are many families who have a child with microtia, but no one on either side of the family has ever had such a child. It appears to be a random event or anomaly occurring during early development. In the case of twins, one child may be born with microtia, but his twin brother will not.
Yes:
Microtia is observed in family generations. Sometimes, an aunt or uncle or cousin has microtia and a new family member is born with the same defect. Relatives may have the right ear affected, and then another family member may be born with a defect in the left ear. Or one family member may have bilateral microtia while the rest have unilateral microtia.
How will microtia affect my baby?
Microtia affects only external hearing.
Microtia may be accompanied by atresia (absence of the ear canal and often hearing loss), hemifacial microsomia, or other syndromes or genetic diseases such as Goldenhar syndrome or Treacher Collins syndrome. However, microtia does not affect a child's intelligence, motor skills, or success in life. Many parents of children with microtia are shocked at first, but soon realize how lucky they are because there are other children in our world who are born with life-threatening conditions. Microtia affects only the ear itself. Children can have different attitudes towards their ears, some don’t mind such ears, others worry. Try to encourage your child on those days when someone teases him about his ear, and the baby doubts his own confidence and feels upset. Help your child understand that he is no different from other children and can get whatever he wants from life.
Developmental anomalies of the external ear
The most common anomalies primarily concern the auricle. Such congenital pathologies are distinguishable visually. They are easily detected when examining a baby not only by doctors, but also by the child’s parents.
Anomalies in the development of the auricle can be divided into:
- those in which the shape of the auricle is modified;
- those in which its dimensions change.
Most often, congenital pathologies combine to varying degrees both a change in the shape and a change in the size of the auricle.
The change in size may be in the direction of enlarging the auricle. This pathology is called macrotia. Microtia
called reduction in the size of the ear.
A change in the size of the auricle until it completely disappears is called anotia
.
The most common defects with changes in the shape of the auricle are as follows:
- The so-called "macaque ear"
. At the same time, the curls in the auricle are smoothed out, almost reduced to nothing. The upper part of the auricle is directed inward; - Prominent ears.
Ears with this malformation have a protruding appearance. Normally, the ears are located parallel to the temporal bone. With protruding ears, they are at an angle to it. The greater the angle of deviation, the greater the degree of protruding ears. When the auricles are located at a right angle to the temporal bone, the protruding ear defect is expressed to the maximum extent. Today, about half of newborns have protruding ears of greater or lesser severity; - The so-called "satyr's ear"
. In this case, the upward stretching of the auricle is pronounced. In this case, the upper tip of the shell has a pointed structure; - Congenital aplasia of the auricle
, also called anotia, is a partial or complete absence of the auricle on one or both sides . It is more common in children with a number of genetic diseases - such as branchial arch syndrome, Goldenhar syndrome and others. Children whose mothers suffered from viral infectious diseases during pregnancy can also be born with anotia.
Aplasia of the auricle can manifest itself in the presence of a small formation of skin-cartilaginous tissue or in the presence of only the lobe. The auditory canal in this case is very narrow. Fistulas can form in parallel in the parotid region. With absolute anotia, that is, the complete absence of the auricle, the ear canal is completely overgrown. The child cannot hear anything with this organ. Surgery is necessary to free the ear canal.
In addition, there are such anomalies as skin growths on them in the form of processes of various shapes. The most acceptable age for children to undergo surgery for ear anomalies is from five to seven years.
Surgical options for ear reconstruction for microtia
1. Using your own costal cartilage.
Reconstructive surgery using rib cartilage has been performed since the 1920s. It is a three to four step procedure that involves the use of costal cartilage (rib graft) and a flap of skin to cover the ear (skin graft). Surgery time for each stage can vary from one hour to five hours.
Because a rib ear graft is made from living body tissue, the ear will grow with the person. The reconstructed ear will be slightly stiffer than a normal ear because the costal cartilage tissue is denser, although to the human eye the difference is almost undetectable. The new ear will feel pain, it will bleed when damaged, and it will heal just like the real one.
2. Medpor surgical technique.
In this case, instead of costal cartilage, frames made of porous Medpor polyethylene are used to reconstruct the auricle. This material has been used for craniomaxillofacial operations for more than forty years. Medpor surgery can be performed in one stage, the surgery time varies from eight to twelve hours depending on the complexity of the operation. The Medpor ear is more rigid than the ear with a rib graft. Blood vessels are integrated into the porous openings of the Medpor frame, allowing the reconstructed ear to feel pain, bleed, and heal.
Reconstructive otoplasty in the absence of a lobe or ear
Defects in the development of the auricles, such as the absence of a lobe or outer ear, require complex reconstructive plastic surgery. This is the most time-consuming and labor-intensive option of otoplasty, which places high demands on the qualifications of the plastic surgeon and is carried out in several stages.
Particularly difficult is the complete reconstruction of the outer ear in the case of its congenital absence (anotia) or loss due to injury. The process of recreating the missing ear is carried out in 3-4 stages and takes about a year.
The first stage includes the formation of the cartilaginous frame of the future ear from the patient’s costal cartilages. At the second stage, the automaterial (cartilaginous base) is placed in a specially formed subcutaneous pocket in place of the missing ear. The implant should take root in its new location within 2-6 months.
During the third stage, the cartilaginous base of the future ear is disconnected from the adjacent tissues of the head, moved to the required position and fixed in the correct position. The wound in the area behind the ear is covered with a skin graft taken from the patient himself (from the arm, leg or abdomen).
And although reconstructive otoplasty cannot restore hearing, the new ear created by surgeons allows patients to experience themselves and the world around them in a new way. The shape of the ear created in the process of reconstructive otoplasty is practically no different from the natural one.
Carrying out reconstructive otoplasty in children who have no external ear is possible no earlier than 6-7 years of age. In case of bilateral hearing loss, early hearing protection (wearing a hearing aid) is indicated so that there is no delay in mental and speech development.
In some cases, with bilateral hearing defects, surgical intervention is performed on the inner ear. An alternative way to solve the cosmetic problem of the absence of an external ear, which is widespread abroad, is to wear a specially made removable auricular prosthesis.
In the absence of an earlobe, operations to restore it are also performed. For this purpose, skin grafts taken from the behind-the-ear space or neck area are used. If such an operation is performed competently and technically, postoperative scars are practically invisible.
Despite the successes achieved by reconstructive plastic surgery in solving the problem of the absence of the lobe and outer ear, the search for new materials and methods of otoplasty continues at present for the most natural reconstruction of such a complex organ in shape and function as the auricle.
According to the World Health Organization, up to 15% of children are born with obvious signs of various developmental abnormalities. However, congenital anomalies can appear later, so in general the incidence of malformations is much higher. It has been established that in children born to older mothers, anomalies occur more often, since the older the woman, the greater the amount of harmful effects of the external environment (physical, chemical, biological) on her body.
Congenital malformations of the outer and middle ear occur with a frequency of 1-2 cases per 10,000 newborns.
The inner ear appears already in the fourth week of embryonic development. The middle ear develops later, and by the time the baby is born, the tympanic cavity contains jelly-like tissue, which subsequently disappears. The outer ear appears in the fifth week of intrauterine development.
In a newborn, the auricle can be enlarged (hypergenesis, macrotia) or reduced (hypogenesis, microtia), which is usually combined with closure of the external auditory canal. Only some of its parts (for example, the earlobe) can be excessively enlarged or reduced.
Developmental anomalies can be unilateral or bilateral and manifest themselves in the form of ear appendages, several auricles (poliotia). There are cleft lobes, congenital ear fistulas, and atresia (absence) of the external auditory canal. The auricle may be absent or occupy an unusual place.
The auricle can be rolled, flat, ingrown, corrugated, angular (macaque ear), pointed (satyr ear). The auricle may have a transverse cleft, and the lobe may have a longitudinal cleft. Other defects of the lobe are also known: it can be adherent, large, or lagging.
Combined forms of external ear defects are not uncommon. Anomalies in the development of the auricle and the external auditory canal are often combined in the form of its partial underdevelopment or complete absence. Such anomalies are described as syndromes. Thus, a malformation of connective tissue, in which many organs are affected, including the ears, is called Marfan syndrome.
For macrotia (increase in the size of the auricle), taking into account the variety of changes, a number of surgical interventions have been proposed. If, for example, the auricle is enlarged evenly in all directions, i.e., has an oval shape, excess tissue can be excised.
Operations to restore the auricle in the absence of it are quite complex because skin is needed, and it is necessary to create an elastic skeleton (support) around which the auricle is formed. To form the skeleton of the auricle, rib cartilage, cartilage of the auricle of a corpse, bone and synthetic materials are used. The ear pendants located near the auricle are removed along with the cartilage.
Congenital anomalies of ear development occur primarily in its outer and middle sections. This is explained by the fact that the elements of the inner and middle ear develop at different times and in different places, so in case of severe congenital anomalies of the outer or middle ear, the inner ear may turn out to be completely normal.
According to domestic and foreign experts, per 10,000 population there are 1-2 cases of congenital anomalies of the external and middle ear (S.N. Lapchenko, 1972). Teratogenic factors are divided into endogenous (genetic) and exogenous (ionizing radiation, drugs, vitamin A deficiency, viral infections - measles rubella, measles, chickenpox, influenza).
Possible damage to: 1) the auricle; 2) auricle, external auditory canal, tympanic cavity; 3) external, middle ear and facial bone defect.
The following malformations of the auricle are observed: macrotia - large auricle; microtia (microtia) - small deformed auricle; anotia (anotia) - absence of the auricle; protruding ears; appendages of the auricle (single or multiple) - small skin formations located in front of the auricle and consisting of skin, subcutaneous fatty tissue and cartilage; parotid (paraauricular) fistulas - a violation of the processes of closing ectodermal pockets (2-3 cases per 1000 newborns), typical localization - the base of the helix, and atypical placement of a paraauricular fistula is possible.
Anomalies of the auricle lead to a cosmetic defect of the face, often combined with underdevelopment or absence of the external auditory canal (Fig. 51, 52, 53). Microtia and underdevelopment of the external auditory canal can be combined with hypoplasia of the entire middle ear.
Rice. 51. Protruding ears
Rice. 52. Microtia and agenesis of the external auditory canal
Rice. 53. Microtia and ear appendages of the auricle
Anomalies in the development of the external auditory canal and middle ear cause conductive hearing loss.
Treatment of congenital anomalies of the outer and middle ear is surgical and is aimed at eliminating the cosmetic defect and reconstructing the sound conducting system of the outer and middle ear. Restoration of the external auditory canal is carried out in children under the age of 7 years, and correction of a cosmetic defect of the auricle is performed closer to 14 years.
Treatment of duck appendages is surgical. They are cut off at the base.
Paraauricular fistulas by themselves do not cause any discomfort (Fig. 54). Only infection and suppuration indicate their presence and require surgical intervention. After opening the abscess and eliminating the purulent process, the epidermal tract is completely removed. Opening the abscess is only a temporary help, since relapses of suppuration are possible in the future.
Currently, congenital malformations of various organs have begun to appear with increased frequency, with which doctors of almost any specialty have to deal.
Moreover, complications can be both purely cosmetic and functional in nature.
Often, various pathologies are accompanied by damage to the hearing organs, and this, in turn, entails disturbances in the child’s psychosomatic state and complicates the development of the speech apparatus.
Moreover, if the lesion is bilateral, it can lead to disability. Since there are many variants of combined developmental anomalies in a child, complex multicomponent treatment is often necessary to cure or alleviate the patient’s condition, requiring certain knowledge from specialists in different fields.
External signs of pathologies in the development of the hearing system can manifest themselves in a variety of ways. The size of the auricle can range from excessively enlarged, as with macrotia, to insignificant or completely absent, as with microtia or anotia. Growths may form in the area of the auricle, for example ear pendants or ear fistulas.
The most serious developmental pathologies leading to complete or partial hearing loss include: atresia or stenosis of the external auditory opening, disorders of the development of the auditory ossicles or labyrinth.
Ear prosthetics
An ear prosthesis is an excellent option that does not require complex surgery. The prosthetic ears look amazing and very real! They can be attached using magnets or glue. You can swim and sleep with dentures. If you decide to reconstruct your ear using an auricle prosthesis made of silicone materials, contact Istok Audio Med LLC
. We cooperate with leading medical institutes and clinics throughout Russia.
119526, Moscow, Vernadsky Ave., 105, bldg. 2. Tkachenko Oksana ext. 525, mobile, Maria Klimacheva ext. 555, mobile,
For a more specific conversation about your case, please send:
- Photo
- Computed Tomography Data
- A copy of the IPR (if there is a disability)
What else can you do?
Nothing to do.
You always have the choice to do nothing and accept your microtia ear as it is, to love your ear. In fact, some people with microtia love their small ears and highlight them with piercings and short hair. Some even believe that their small ears are a distinctive feature, something that makes them stand out from the crowd. Visit foreign sites where you will find a lot of useful information about microtia:
In Russia, active communication on microtia issues is conducted on the VKontakte network, the microtia group https://vk.com/microtia
The article was written using materials from the sites: